This error troubled me a lot while parsing some data, at last this error is resolved for my case.
filename: test_genome_lengths:
KP5_Contig1 1099843
KP5_Contig2 939199
KP5_Contig3 804334
KP5_Contig4 704755
KP5_Contig5 490858
KP5_Contig6 445261
KP5_Contig7 336421
KP5_Contig8 205120
KP5_Contig9 173756
KP5_Contig10 63375
KP5_Contig11 4752
filename: original.bed
KP5_Contig1 2 378871
KP5_Contig1 378872 812978
KP5_Contig1 814316 1099843
KP5_Contig10 27093 28206
KP5_Contig10 30740 42583
KP5_Contig10 43383 46800
KP5_Contig10 47283 51877
KP5_Contig10 52485 57209
KP5_Contig10 57496 57838
KP5_Contig11 1 902
KP5_Contig11 3859 4197
KP5_Contig11 4429 4752
KP5_Contig2 1 939199
KP5_Contig3 1 8672
bedtools complement -i original.bed -g test_genome_lengths
Error: Sorted input specified, but the file has the following out of order record with a different sort order than the genomeFile
KP5_Contig2 1 939199
This is caused because my bed file is not sorted numerically using sort. The correct order I needed to input was:
filename: corrected.bed
KP5_Contig1 2 378871
KP5_Contig1 378872 812978
KP5_Contig1 814316 1099843
KP5_Contig2 1 939199
KP5_Contig3 1 8672
KP5_Contig10 27093 28206
KP5_Contig10 30740 42583
KP5_Contig10 43383 46800
KP5_Contig10 47283 51877
KP5_Contig10 52485 57209
KP5_Contig10 57496 57838
KP5_Contig11 1 902
KP5_Contig11 3859 4197
KP5_Contig11 4429 4752
bedtools complement -i
corrected.bed -g test_genome_lengths
Now, no error exists.
To get corrected.bed, I have sorted numerically in the following way:
cat original.bed | sort -n -k1.11 -nk2,2 >corrected.bed
For more info on this sort function, check this stack overflow post.
Tuesday, November 20, 2018
Thursday, September 27, 2018
Downloading fasta using efetch - CentOS
$ efetch -db nucleotide -id KJ413946.1 -format fasta 501 Protocol scheme 'https' is not supported (LWP::Protocol::https not installed) No do_post output returned from 'https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=nucleotide&id=KJ413946.1&rettype=fasta&retmode=text&edirect_os=linux&edirect=9.90&tool=edirect&email=datta@ttshbio' Result of do_post http request is $VAR1 = bless( { '_content' => 'LWP will support https URLs if the LWP::Protocol::https module is installed. ', '_rc' => 501, '_headers' => bless( { 'client-warning' => 'Internal response', 'client-date' => 'Thu, 27 Sep 2018 06:42:06 GMT', 'content-type' => 'text/plain', '::std_case' => { 'client-warning' => 'Client-Warning', 'client-date' => 'Client-Date' } }, 'HTTP::Headers' ), '_msg' => 'Protocol scheme \'https\' is not supported (LWP::Protocol::https not installed)', '_request' => bless( { '_content' => 'db=nucleotide&id=KJ413946.1&rettype=fasta&retmode=text&edirect_os=linux&edirect=9.90&tool=edirect&email=datta@ttshbio', '_uri' => bless( do{\(my $o = 'https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi')}, 'URI::https' ), '_headers' => bless( { 'user-agent' => 'libwww-perl/6.05', 'content-type' => 'application/x-www-form-urlencoded' }, 'HTTP::Headers' ), '_method' => 'POST' }, 'HTTP::Request' ) }, 'HTTP::Response' );
Installing "perl-LWP-Protocol-https" has solved the problem!
$ sudo yum install perl-LWP-Protocol-https
$ efetch -db nucleotide -id KJ413946.1 -format fasta >KJ413946.1 Escherichia coli strain ECS01 plasmid pNDM-ECS01, complete sequence ATGGCAGAGGAAAGCAAACAGCTAACCAAACGGCAACAAAAAGCCATTGATACAGCGGCGTTAATCCGGC AGGAGCCGCCGCAGGGTGAAGATATGGCATTCACCCACTCCATTCTGTGCCAGGTCGGTTTGCCCCGTTC TAAGGTGGCAGGGCGTGAGTTTATGCGCCGTTCTGGTGATGCCTGGCTCGTCGTACAGGCAGGCTGGATT GATGAAGGCAGTGGCCCGGTAGAGCAGCCTTTACCCTATGGCGCTATGCCGCGACTCACGTTCGCCTGGA TTTCATCGTATGCACTGCGCAACAAAACGCGGGAAATCGCCATCGGCCACAGCGCTAATGAGTTTCTTCA CCTTATGGGGATGGACTCACAGGGAACCCGTCATAAAACGCTGCGTACACAAATGCAGGCGCTGGCCGCG TGTCGTTTGCAGCTGGGCTTTAAGGGCC
$ time for d in $(cat Plasmid.list);do echo $d; efetch -db nucleotide -id $d -format fasta >$d.fasta; done ##takes a bit longer time real 6m47.812s
user 0m43.515s
sys 0m7.811s For extracting multiple records from local NT/NR database with a list of accessions using "blastdbcmd" and make accession as filename
$ time for d in $(cat Plasmid.list);do echo $d; blastdbcmd -db nt -entry $d >$d.fasta; done
real 0m26.736s
user 0m7.004s
sys 0m4.384s
Notice the advantage of having downloaded local databases. It just took 26 secs!!
Wednesday, September 26, 2018
Cannot find xml2-config - R Studio - CentOS
Came across this error while installing CINNA package on CentOS.
On terminal:
then in R:
install.packages("XML")
This worked without error!
checking for xml2-config... no Cannot find xml2-config ERROR: configuration failed for package ‘XML’ * removing ‘/home/datta/R/x86_64-redhat-linux-gnu-library/3.4/XML’ Warning in install.packages : installation of package ‘XML’ had non-zero exit status
On terminal:
$ sudo yum install libxml2-devel Loaded plugins: fastestmirror, langpacks Loading mirror speeds from cached hostfile
* base: centos.netonboard.com * epel: mirror.premi.st * extras: mirror.myren.net.my * ius: hkg.mirror.rackspace.com * nux-dextop: mirror.li.nux.ro * updates: centos.ipserverone.com Resolving Dependencies --> Running transaction check ---> Package libxml2-devel.x86_64 0:2.9.1-6.el7_2.3 will be installed --> Finished Dependency Resolution Dependencies Resolved ................... Installed: libxml2-devel.x86_64 0:2.9.1-6.el7_2.3 Complete!
then in R:
install.packages("XML")
This worked without error!
Sunday, September 23, 2018
makeblastdb - Segmentation fault (core dumped)
I had a list of sequence which I wanted to use as NCBI BLAST database. makeblastdb (version 2.7.1+) was throwing me the following error!
Tried the following:
1) Let's remove comma's
2) Now let's try removing brackets in fasta headers
3. Shorten the header - This has worked!!
Appears like makeblastdb has a limit for the length for the headers in fasta. Should take note of it!
$ grep '>' NCBI_PlasmidCluster_TophitsSeqs.fasta
>NZ_CM008903.1_Enterobacter_cloacae_strain_22ES_plasmid_p22ES-4970,_whole_genome_shotgun_sequence >NZ_CM004622.1_Escherichia_coli_strain_Ec47VL_plasmid_pEC47a,_whole_genome_shotgun_sequence >KC355363.1_Aeromonas_hydrophila_strain_AH1_plasmid_pN6,_partial_sequence >HQ651092.1_Klebsiella_oxytoca_plasmid_pFP10-1_replication_protein_gene,_partial_cds;_hypothetical_protein_(pKP048_08),_antirestriction_protein_KlcA_(klcA),_hypothetical_protein_(pKP048_09),_transcriptional_repressor_protein_KorC_(korC),_putative_ISKpn6-like_transposase_(tnpA),_and_carbapenemase_KPC-2_(blaKPC-2)_genes,_complete_cds;_and_transposon_Tn3_truncated_TEM-beta-lactamase_(bla)_and_ISKpn8_transposase_(tnpA)_genes,_complete_cds,_insertion_sequence_ISKpn8,_complete_sequence,_and_TnpR_(tnpR)_gene,_partial_cds $ makeblastdb -in NCBI_PlasmidCluster_TophitsSeqs.fasta -dbtype nucl -out NCBI_PlasmidCluster_TophitsSeqs.fasta.DB -parse_seqids Building a new DB, current time: 09/24/2018 11:02:13 New DB name: NCBI_PlasmidCluster_TophitsSeqs.fasta.DB New DB title: NCBI_PlasmidCluster_TophitsSeqs.fasta Sequence type: Nucleotide Keep MBits: T Maximum size: 1000000000B CFastaReader: Near line 1, the sequence id string contains 'comma' symbol, which has been replaced with 'underscore' symbol. Please correct the sequence id string. CFastaReader: Near line 74, the sequence id string contains 'comma' symbol, which has been replaced with 'underscore' symbol. Please correct the sequence id string. CFastaReader: Near line 132, the sequence id string contains 'comma' symbol, which has been replaced with 'underscore' symbol. Please correct the sequence id string. CFastaReader: Near line 298, the sequence id string contains 'comma' symbol, which has been replaced with 'underscore' symbol. Please correct the sequence id string. Adding sequences from FASTA; added 4 sequences in 0.00256801 seconds. Segmentation fault (core dumped)
Tried the following:
1) Let's remove comma's
$ sed 's/,/_/g' NCBI_PlasmidCluster_TophitsSeqs.fasta >NCBI_PlasmidCluster_TophitsSeqs_1NoCommas.fasta $ grep '>' NCBI_PlasmidCluster_TophitsSeqs_1NoCommas.fasta >NZ_CM008903.1_Enterobacter_cloacae_strain_22ES_plasmid_p22ES-4970__whole_genome_shotgun_sequence >NZ_CM004622.1_Escherichia_coli_strain_Ec47VL_plasmid_pEC47a__whole_genome_shotgun_sequence >KC355363.1_Aeromonas_hydrophila_strain_AH1_plasmid_pN6__partial_sequence >HQ651092.1_Klebsiella_oxytoca_plasmid_pFP10-1_replication_protein_gene__partial_cds;_hypothetical_protein_(pKP048_08)__antirestriction_protein_KlcA_(klcA)__hypothetical_protein_(pKP048_09)__transcriptional_repressor_protein_KorC_(korC)__putative_ISKpn6-like_transposase_(tnpA)__and_carbapenemase_KPC-2_(blaKPC-2)_genes__complete_cds;_and_transposon_Tn3_truncated_TEM-beta-lactamase_(bla)_and_ISKpn8_transposase_(tnpA)_genes__complete_cds__insertion_sequence_ISKpn8__complete_sequence__and_TnpR_(tnpR)_gene__partial_cds $ makeblastdb -in NCBI_PlasmidCluster_TophitsSeqs_1NoCommas.fasta -dbtype nucl -out NCBI_PlasmidCluster_TophitsSeqs.fasta.DB -parse_seqids Building a new DB, current time: 09/24/2018 11:08:16 New DB name: NCBI_PlasmidCluster_TophitsSeqs.fasta.DB New DB title: NCBI_PlasmidCluster_TophitsSeqs_1NoCommas.fasta Sequence type: Nucleotide Keep MBits: T Maximum size: 1000000000B Adding sequences from FASTA; added 4 sequences in 0.00210285 seconds. Segmentation fault (core dumped)
2) Now let's try removing brackets in fasta headers
$ sed 's/[()]/_/g' NCBI_PlasmidCluster_TophitsSeqs_1NoCommas.fasta >NCBI_PlasmidCluster_TophitsSeqs_2NoBrackets.fasta $ makeblastdb -in NCBI_PlasmidCluster_TophitsSeqs_2NoBrackets.fasta -dbtype nucl -out NCBI_PlasmidCluster_TophitsSeqs.fasta.DB -parse_seqids Building a new DB, current time: 09/24/2018 11:11:39 New DB name: NCBI_PlasmidCluster_TophitsSeqs.fasta.DB New DB title: NCBI_PlasmidCluster_TophitsSeqs_2NoBrackets.fasta Sequence type: Nucleotide Keep MBits: T Maximum size: 1000000000B
Adding sequences from FASTA; added 4 sequences in 0.00201416 seconds. Segmentation fault (core dumped)
3. Shorten the header - This has worked!!
$ cat NCBI_PlasmidCluster_TophitsSeqs_2NoBrackets.fasta | cut -f1 -d ";" >NCBI_PlasmidCluster_TophitsSeqs_3_Shortened.fasta $ makeblastdb -in NCBI_PlasmidCluster_TophitsSeqs_3_Shortened.fasta -dbtype nucl -out NCBI_PlasmidCluster_TophitsSeqs.fasta.DB -parse_seqids Building a new DB, current time: 09/24/2018 11:19:48 New DB name: NCBI_PlasmidCluster_TophitsSeqs.fasta.DB New DB title: NCBI_PlasmidCluster_TophitsSeqs_3_Shortened.fasta Sequence type: Nucleotide Keep MBits: T Maximum size: 1000000000B Adding sequences from FASTA; added 4 sequences in 0.00199008 seconds.
Appears like makeblastdb has a limit for the length for the headers in fasta. Should take note of it!
Wednesday, September 19, 2018
BLASTN Error: NCBI C++ Exception
Ran into this error today morning.
Found that incomplete or corrupted database causes this problem. I had two copies of the same NT database, so I ran md5sum for each of the file in the database and found the md5sum of one of the file was not same. (see below)

the long string above is md5sum,file name. - Note the md5sums of the same file are not same.
So just copied the nt.00.nhr file from the archive and blast worked without trouble. If not, you have to download the database again or run makeblastdb again on the fasta sequences.
Error: NCBI C++ Exception: T0 "/home/coremake/release_build/build/PrepareRelease_Linux64-Centos_JSID_01_380670_130.14.18.6_9008__PrepareRelease_Linux64-Centos_1508370803/c++/compilers/unix/../../src/serial/objistrasnb.cpp", line 179: Error: ncbi::CObjectIStreamAsnBinary::UnexpectedTagClassByte() - byte 0: unexpected tag: application/constructed/11 (107), should be constructed/None (32) ( at Blast-def-line-set[])
Found that incomplete or corrupted database causes this problem. I had two copies of the same NT database, so I ran md5sum for each of the file in the database and found the md5sum of one of the file was not same. (see below)
the long string above is md5sum,file name. - Note the md5sums of the same file are not same.
So just copied the nt.00.nhr file from the archive and blast worked without trouble. If not, you have to download the database again or run makeblastdb again on the fasta sequences.
Sunday, August 26, 2018
Matching alphanumeric string in PERL
if($_ =~ /[a-zA-Z]+/ && $_ =~ /[0-9]+/)
$_ : the string in default variable
=~ : should match
a-zA-Z: lower case and uppercase
+ : more than once
&&: as well as
0-9: numbers
Connection timed out; no servers could be reached - CentOS - Solved
For some reason, could not connect access internet in CentOS, although my network shows connected. $ nslookup google.com ;; connection timed out; no servers could be reached $ ping www.google.com ping: www.google.com: Name or service not known $ ping 8.8.8.8 From XX.XX.X.X icmp_seq=1 Destination Net Unreachable From XX.XX.X.X icmp_seq=2 Destination Net Unreachable From XX.XX.X.X icmp_seq=3 Destination Net Unreachable From XX.XX.X.X icmp_seq=4 Destination Net Unreachable
then, from an online post, I understand that/etc/sysconfig/network should have following:
$ cat /etc/sysconfig/network
NETWORKING=yes
When I checked my/etc/sysconfig/network, it was empty. Soon, I updated it withNETWORKING=yes GATEWAY=XX.XX.X.X (filled with my IP address)and restarted my system again, which works now without problem!!
Wednesday, August 22, 2018
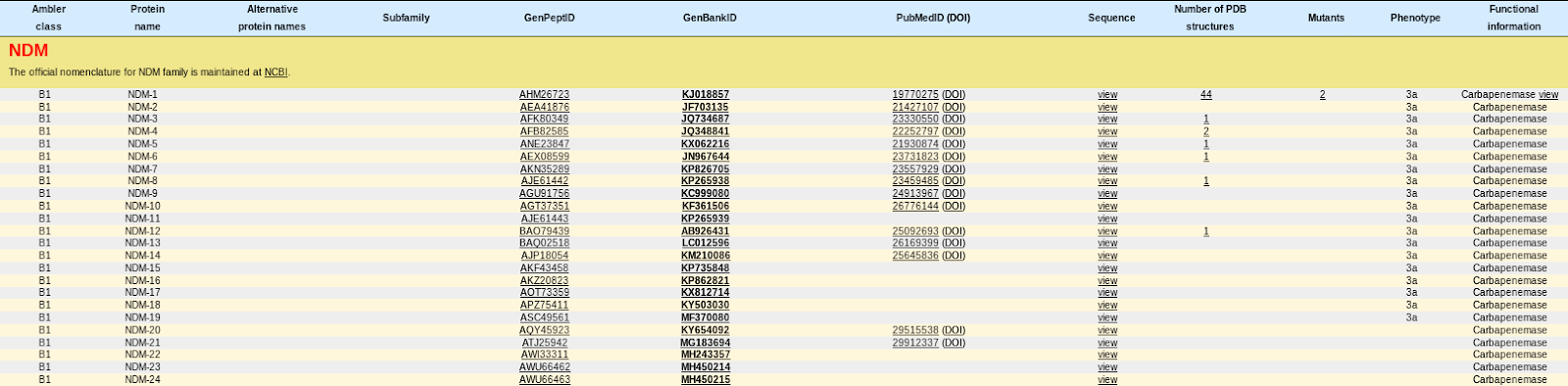
List of carbapenamase genes available till date
can find more CP genes at this webpage: http://bldb.eu/BLDB.php?prot=B1
Sunday, August 19, 2018
Gene Ontology information from Uniport IDs
1) Go to https://www.uniprot.org/uploadlists/, paste your identifiers and submit

2) Once you get results, click Gene Ontology on the left side to gene ontology annotations.

3) Check the results by clicking the "+" button. Once it expands you can see more details

4) Once it expands you can see more details like below:

Thursday, August 9, 2018
Generate combinations from a item list in BASH + Ignore same pairs
View the contents of the file using cat command
A bash "for" loop in another "for" helps + AWK resolves the issue
$ cat list Pair1 Pair2 Pair3 Pair4
A bash "for" loop in another "for" helps + AWK resolves the issue
$ for d in $(cat list); do for i in $(cat list); do echo "$d $i"; done done | awk '$1 != $2' Pair1 Pair2 Pair1 Pair3 Pair1 Pair4 Pair2 Pair1 Pair2 Pair3 Pair2 Pair4 Pair3 Pair1 Pair3 Pair2 Pair3 Pair4 Pair4 Pair1 Pair4 Pair2 Pair4 Pair3
Sequence lengths from AWK one-liner
$ awk '/^>/ {if (sqlen){print sqlen}; print ;sqlen=0;next; } { sqlen = sqlen +length($0)}END{print sqlen}' db4_cdc_NDM_only.fasta | sed 's/>//g' | paste - - ref|NZ_CP029245.1| Escherichia_coli_strain_ECCRA-119_plasmid_pTB203,_complete_sequence 46161 ref|NZ_CP029386.1| Klebsiella_pneumoniae_subsp._pneumoniae_strain_SCKP040074_plasmid_pNDM6_040074,_complete_sequence 52989 ref|NZ_CP029731.1| Citrobacter_sp._CRE-46_strain_AR_0157_plasmid_unnamed3,_complete_sequence 108148 ref|NZ_CP029737.1| Providencia_rettgeri_strain_AR_0082_plasmid_unnamed,_complete_sequence 144970 ref|NZ_CP029978.1| Escherichia_coli_strain_51008369SK1_plasmid_p51008369SK1_E,_complete_sequence 99465 ref|NZ_KM923969.1| Acinetobacter_dijkshoorniae_strain_JVAP01_plasmid_pNDM-JVAP01,_complete_sequence 47268
Friday, August 3, 2018
Setting up AWS CLI and Downloading S3 Files using command line interface
1. Install the AWS Command Line Interface (CLI) using:
For more details such as upgrade, check the amazon webpage here.
Note: if installation was not successful, reinstall forcefully. Check here
The next step is to configure. AWS requires the following items for this:
(how to find these?)
Now, run the following command and answer the prompts:
3. Copying files from AWS S3 folder to Local Desktop
once this is done, we can start using AWS CLI for the file operations. For example, if it want to copy WBB3097.gz file in my AWS account to my local desktop, which is under "qgen" username and in the folder MIX7341, I do the following
4. List all files in AWS S3 folder
Similarly to list all the files in the MIX7341, we can use the following command
5. Copy multiple files using BASH for loop
Off hand, I had to download multiple files from AWS account. To click each file and download was taking time. So, I just listed the files (step1 below) and used "for loop" in bash to download the files (step2 below).
border:solid green;border-width:.1em .1em .1em .8em;padding:.2em .6em;
Python ; Pastie ; www. hilitie.me
$ pip install awscli
For more details such as upgrade, check the amazon webpage here.
Note: if installation was not successful, reinstall forcefully. Check here
The next step is to configure. AWS requires the following items for this:
(how to find these?)
- Access key ID
- Secret Access key
- Region name
- Valid output format
Now, run the following command and answer the prompts:
$ aws configure
AWS Access Key ID [None]: <enter the access key> AWS Secret Access Key [None]: <enter the secret access key> Default region name [None]: <enter region> Default output format [None]: <text>
3. Copying files from AWS S3 folder to Local Desktop
once this is done, we can start using AWS CLI for the file operations. For example, if it want to copy WBB3097.gz file in my AWS account to my local desktop, which is under "qgen" username and in the folder MIX7341, I do the following
$ aws s3 cp s3:URI <Local Desktop Location> $ aws s3 cp s3://qgen/MIX7341/WBB3097.gz ~/Desktop/AWS
4. List all files in AWS S3 folder
Similarly to list all the files in the MIX7341, we can use the following command
$ aws s3 ls s3://qgen/MIX7341/ 2018-08-02 11:03:58 459407360 WBB3066.gz 2018-08-02 11:03:01 337848320 WBB3067.gz 2018-08-02 11:02:57 327219200 WBB3068.gz 2018-08-02 11:02:57 298680320 WBB3069.gz 2018-08-02 11:02:57 280514560 WBB3070.gz
5. Copy multiple files using BASH for loop
Off hand, I had to download multiple files from AWS account. To click each file and download was taking time. So, I just listed the files (step1 below) and used "for loop" in bash to download the files (step2 below).
$ aws s3 ls s3://qgen/MIX7341/ | awk '{print $NF}' >files_in_AWS.list ##step 1 $ for d in $(cat files_in_AWS.list); do echo $d; aws s3 cp s3://qgen/MIX7341/$d . ; done ##step 2
border:solid green;border-width:.1em .1em .1em .8em;padding:.2em .6em;
Python ; Pastie ; www. hilitie.me
Thursday, August 2, 2018
ImportError: No module named botocore.session - Solved
My aws cli had some installation problems and ended up using the following error
Running the following command fixed the problem:
We can check aws version as following:
Thats it ! We can now use AWS CLI to access the buckets in our AWS console account. :)
$ aws Traceback (most recent call last): File "/usr/bin/aws", line 19, in <module> import awscli.clidriver File "/usr/lib/python2.7/site-packages/awscli/clidriver.py", line 17, in <module> import botocore.session ImportError: No module named botocore.session
Running the following command fixed the problem:
$ sudo pip install awscli --force-reinstall --upgrade Collecting awscli Using cached https://files.pythonhosted.org/packages/18/99/a1a1ea5a91161d5be5f434550ac1de800d79da7d4f68a5b5c8f265fcbd58/awscli-1.15.70-py2.py3-none-any.whl Collecting s3transfer<0.2.0,>=0.1.12 (from awscli) Using cached https://files.pythonhosted.org/packages/d7/14/2a0004d487464d120c9fb85313a75cd3d71a7506955be458eebfe19a6b1d/s3transfer-0.1.13-py2.py3-none-any.whl Collecting colorama<=0.3.9,>=0.2.5 (from awscli) Using cached https://files.pythonhosted.org/packages/db/c8/7dcf9dbcb22429512708fe3a547f8b6101c0d02137acbd892505aee57adf/colorama-0.3.9-py2.py3-none-any.whl Collecting rsa<=3.5.0,>=3.1.2 (from awscli) Using cached https://files.pythonhosted.org/packages/e1/ae/baedc9cb175552e95f3395c43055a6a5e125ae4d48a1d7a924baca83e92e/rsa-3.4.2-py2.py3-none-any.whl Collecting docutils>=0.10 (from awscli) Using cached https://files.pythonhosted.org/packages/50/09/c53398e0005b11f7ffb27b7aa720c617aba53be4fb4f4f3f06b9b5c60f28/docutils-0.14-py2-none-any.whl Collecting PyYAML<=3.13,>=3.10 (from awscli) Downloading https://files.pythonhosted.org/packages/9e/a3/1d13970c3f36777c583f136c136f804d70f500168edc1edea6daa7200769/PyYAML-3.13.tar.gz (270kB) 100% |████████████████████████████████| 276kB 2.8MB/s Collecting botocore==1.10.69 (from awscli) Using cached https://files.pythonhosted.org/packages/45/f3/0e5006bc6198213b853ca3975c537335a91716ffa1ed768e2b2ffe12d7ed/botocore-1.10.69-py2.py3-none-any.whl Collecting futures<4.0.0,>=2.2.0; python_version == "2.6" or python_version == "2.7" (from s3transfer<0.2.0,>=0.1.12->awscli) Using cached https://files.pythonhosted.org/packages/2d/99/b2c4e9d5a30f6471e410a146232b4118e697fa3ffc06d6a65efde84debd0/futures-3.2.0-py2-none-any.whl Collecting pyasn1>=0.1.3 (from rsa<=3.5.0,>=3.1.2->awscli) Downloading https://files.pythonhosted.org/packages/d1/a1/7790cc85db38daa874f6a2e6308131b9953feb1367f2ae2d1123bb93a9f5/pyasn1-0.4.4-py2.py3-none-any.whl (72kB) 100% |████████████████████████████████| 81kB 3.5MB/s Collecting jmespath<1.0.0,>=0.7.1 (from botocore==1.10.69->awscli) Using cached https://files.pythonhosted.org/packages/b7/31/05c8d001f7f87f0f07289a5fc0fc3832e9a57f2dbd4d3b0fee70e0d51365/jmespath-0.9.3-py2.py3-none-any.whl Collecting python-dateutil<3.0.0,>=2.1; python_version >= "2.7" (from botocore==1.10.69->awscli) Using cached https://files.pythonhosted.org/packages/cf/f5/af2b09c957ace60dcfac112b669c45c8c97e32f94aa8b56da4c6d1682825/python_dateutil-2.7.3-py2.py3-none-any.whl Collecting six>=1.5 (from python-dateutil<3.0.0,>=2.1; python_version >= "2.7"->botocore==1.10.69->awscli) Using cached https://files.pythonhosted.org/packages/67/4b/141a581104b1f6397bfa78ac9d43d8ad29a7ca43ea90a2d863fe3056e86a/six-1.11.0-py2.py3-none-any.whl Building wheels for collected packages: PyYAML Running setup.py bdist_wheel for PyYAML ... done Stored in directory: /root/.cache/pip/wheels/ad/da/0c/74eb680767247273e2cf2723482cb9c924fe70af57c334513f Successfully built PyYAML ipaclient 4.5.4 requires jinja2, which is not installed. rtslib-fb 2.1.63 has requirement pyudev>=0.16.1, but you'll have pyudev 0.15 which is incompatible. ipapython 4.5.4 has requirement dnspython>=1.15, but you'll have dnspython 1.12.0 which is incompatible. Installing collected packages: futures, jmespath, docutils, six, python-dateutil, botocore, s3transfer, colorama, pyasn1, rsa, PyYAML, awscli Found existing installation: futures 3.2.0 Uninstalling futures-3.2.0: Successfully uninstalled futures-3.2.0 Found existing installation: jmespath 0.9.3 Uninstalling jmespath-0.9.3: Successfully uninstalled jmespath-0.9.3 Found existing installation: docutils 0.14 Uninstalling docutils-0.14: Successfully uninstalled docutils-0.14 Found existing installation: six 1.9.0 Uninstalling six-1.9.0: Successfully uninstalled six-1.9.0 Found existing installation: python-dateutil 2.7.3 Uninstalling python-dateutil-2.7.3: Successfully uninstalled python-dateutil-2.7.3 Found existing installation: botocore 1.10.69 Uninstalling botocore-1.10.69: Successfully uninstalled botocore-1.10.69 Found existing installation: s3transfer 0.1.13 Uninstalling s3transfer-0.1.13: Successfully uninstalled s3transfer-0.1.13 Found existing installation: colorama 0.3.9 Uninstalling colorama-0.3.9: Successfully uninstalled colorama-0.3.9 Found existing installation: pyasn1 0.1.9 Uninstalling pyasn1-0.1.9: Successfully uninstalled pyasn1-0.1.9 Found existing installation: rsa 3.4.2 Uninstalling rsa-3.4.2: Successfully uninstalled rsa-3.4.2 Found existing installation: PyYAML 3.10 Cannot uninstall 'PyYAML'. It is a distutils installed project and thus we cannot accurately determine which files belong to it which would lead to only a partial uninstall. You are using pip version 10.0.1, however version 18.0 is available. You should consider upgrading via the 'pip install --upgrade pip' command. $ aws usage: aws [options] <command> <subcommand> [<subcommand> ...] [parameters] To see help text, you can run: aws help aws <command> help aws <command> <subcommand> help aws: error: too few arguments
We can check aws version as following:
$ aws --version aws-cli/1.15.70 Python/2.7.5 Linux/3.10.0-693.17.1.el7.x86_64 botocore/1.10.69
Thats it ! We can now use AWS CLI to access the buckets in our AWS console account. :)
Tuesday, July 10, 2018
IndexError: string index out of range - Parsnp - Solved
$ parsnp_linux_ignoresize -r ecoli_NZ_CP027462.fasta -d . -o coregenome -x -c
|--Parsnp v1.2--|
For detailed documentation please see --> http://harvest.readthedocs.org/en/latest
***************************
SETTINGS:
|-refgenome: /storage/data/DATA4/analysis/Datta/CRE_outbreak/Trees/Parnsp_Gubbins_Concordant_Samples/CRE_Paola-GIS/ecoli_gte1kbContigs/ecoli_NZ_CP027462.fasta
|-aligner: libMUSCLE
|-seqdir: .
|-outdir: coregenome
|-OS: Linux
|-threads: 32
***************************
<<Parsnp started>>
-->Reading Genome (asm, fasta) files from ...
|->[OK]
-->Reading Genbank file(s) for reference (.gbk) ..
|->[WARNING]: no genbank file provided for reference annotations, skipping..
Traceback (most recent call last):
File "<string>", line 656, in <module>
IndexError: string index out of range
Solution: a previous failed run generated empty parsnp.fasta file which led to this error. By deleting parsnp.fasta fixed the script run again.
|--Parsnp v1.2--|
For detailed documentation please see --> http://harvest.readthedocs.org/en/latest
***************************
SETTINGS:
|-refgenome: /storage/data/DATA4/analysis/Datta/CRE_outbreak/Trees/Parnsp_Gubbins_Concordant_Samples/CRE_Paola-GIS/ecoli_gte1kbContigs/ecoli_NZ_CP027462.fasta
|-aligner: libMUSCLE
|-seqdir: .
|-outdir: coregenome
|-OS: Linux
|-threads: 32
***************************
<<Parsnp started>>
-->Reading Genome (asm, fasta) files from ...
|->[OK]
-->Reading Genbank file(s) for reference (.gbk) ..
|->[WARNING]: no genbank file provided for reference annotations, skipping..
Traceback (most recent call last):
File "<string>", line 656, in <module>
IndexError: string index out of range
Solution: a previous failed run generated empty parsnp.fasta file which led to this error. By deleting parsnp.fasta fixed the script run again.
Wednesday, June 20, 2018
Converting date to fractional date for BEAST phylogenetic analysis software
While running BEAST, we need to generate an xml with the parameters. When we import the alignment file into BEAUti software and we require the sampling time in fractional years. Here is one way to do that in the excel. Regardless of your date is in any of the formats, the formula would convert that into a fractional year.
=YEAR(A1)+((A1)-DATE(YEAR(A1),1,0))/(DATE(YEAR(A1)+1,1,0)-DATE(YEAR(A1),1,0))

=YEAR(A1)+((A1)-DATE(YEAR(A1),1,0))/(DATE(YEAR(A1)+1,1,0)-DATE(YEAR(A1),1,0))
Tuesday, February 13, 2018
Merged entries in the non-redundant Nucleotide (nt) database
I ran the following command for to extract a sequence from nt database:
I noticed that this is a merged entry in the 'non-redundant' Nucleotide (nt) database, where two nucleotide sequences which have the same sequence are combined under a single FASTA entry.
When I used the second identifier in the merged entry, blastdbcmd command gave me the same result in return:


$ blastdbcmd -db /mnt/Storage/nt/nt/nt -entry KJ413946.1 | grep '>' >KJ413946.1 Escherichia coli strain ECS01 plasmid pNDM-ECS01, complete sequence >KP900017.1 Raoultella ornithinolytica strain YNKP001 plasmid pYNKP001-NDM, complete sequence
I noticed that this is a merged entry in the 'non-redundant' Nucleotide (nt) database, where two nucleotide sequences which have the same sequence are combined under a single FASTA entry.
When I used the second identifier in the merged entry, blastdbcmd command gave me the same result in return:
$ blastdbcmd -db /mnt/Storage/nt/nt/nt -entry KP900017.1 | grep '>' >KJ413946.1 Escherichia coli strain ECS01 plasmid pNDM-ECS01, complete sequence >KP900017.1 Raoultella ornithinolytica strain YNKP001 plasmid pYNKP001-NDM, complete sequence
Curious to know, I checked if they are the same length. Yes they are of same length (41190).
$ blastdbcmd -db /mnt/Storage/nt/nt/nt -entry KP900017.1 | grep -v '>' | tr -d '\n' | wc 0 1 41190 $ blastdbcmd -db /mnt/Storage/nt/nt/nt -entry KJ413946.1 | grep -v '>' | tr -d '\n' | wc 0 1 41190
Again curious to know, if these two sequences are sharing an exact similarity match, I blasted both of them. And with no surprise, they share 100 % identity. See blast screenshots below:

So, how to sort this out? I need only one annotation instead of multiple for my analysis and do not want the second identifier to confuse my downstream analysis scripts.
By searching around, problem could solved by using "-target_only" option while running the command. I believe this situation might arise in nr database too but switching this on should help to achieve the purpose.
$ blastdbcmd -db /mnt/Storage/nt/nt/nt -entry KJ413946.1 -target_only | grep '>' >KJ413946.1 Escherichia coli strain ECS01 plasmid pNDM-ECS01, complete sequence
Wednesday, February 7, 2018
[bcf_sync] incorrect number of fields (0 != 5) at 0:0
The following samtools v0.1.18 mpileup command gave me error:
$ samtools mpileup -E -M0 -Q25 -q30 -m2 -D -S test_SortSam_withReadGroup.bam | bcftools view -vcg - > test.vcf
[mpileup] 1 samples in 1 input files
<mpileup> Set max per-file depth to 8000
[bcf_sync] incorrect number of fields (0 != 5) at 0:0
[afs] 0:0.000
including option "-g" helped to overcome the problem.
$ samtools mpileup -g -E -M0 -Q25 -q30 -m2 -D -S test_S8_SortSam_withReadGroup.bam | bcftools view -vcg - > test.vcf
"-g " computes genotype likelihoods and output them in the binary call format (BCF).
$ samtools mpileup -E -M0 -Q25 -q30 -m2 -D -S test_SortSam_withReadGroup.bam | bcftools view -vcg - > test.vcf
[mpileup] 1 samples in 1 input files
<mpileup> Set max per-file depth to 8000
[bcf_sync] incorrect number of fields (0 != 5) at 0:0
[afs] 0:0.000
including option "-g" helped to overcome the problem.
$ samtools mpileup -g -E -M0 -Q25 -q30 -m2 -D -S test_S8_SortSam_withReadGroup.bam | bcftools view -vcg - > test.vcf
"-g " computes genotype likelihoods and output them in the binary call format (BCF).
Thursday, February 1, 2018
One liner to convert a long fasta-file into many separate single fasta sequences
Taken from the Biostar post: https://www.biostars.org/p/105388/
while read line; do if [[ ${line:0:1} == '>' ]]; then outfile=${line#>}.fa; echo $line > $outfile; else echo $line >> $outfile; fi; done < combined_kpneumoniae.tfa
while read line; do if [[ ${line:0:1} == '>' ]]; then outfile=${line#>}.fa; echo $line > $outfile; else echo $line >> $outfile; fi; done < combined_kpneumoniae.tfa
Wednesday, January 10, 2018
Coloring specific tips labels of phylogenetic tree in R

Subscribe to:
Posts (Atom)